FAQ's
TM-Aligner is a protein sequence alignment tool developed in C, Perl and PHP. TM-Aligner uses the progressive alignment strategy for aligning protein sequences and UPGMA method is used to find similar sequences which guide the alignment process. In TM-Aligner, transmembrane regions within the input protein sequences are predicted and aligned using dynamic programming. Cytoplasmic and non-cytoplasmic regions are sieved and aligned using Wu-Manber string matching algorithm.
To limit load to server we have put a limit of 5000 sequences or filesize less than 2MB uploaded to server. If filesize is greater than 2MB please forward it directly to webmaster.
The default transition matrix is Phat, gap opening penalty is 8 bits, gap extension is 1 bit.
This occurs when something is wrong with input sequences either with the format or sequence name.
TM-Aligner assumes every sequences have unique name.
Results are stored at TM-Aligner for a week from the date of job submission.
TM-Aligner color different residues in the basic of their physicochemical properties:
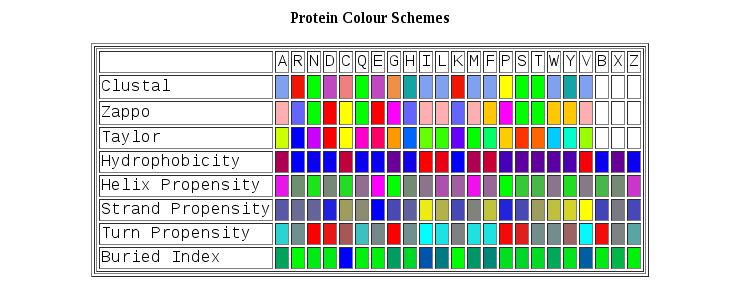
Image taken from http://www.jalview.org/help/html/colourSchemes/
Email your query to bbasharat [at] outlook [dot] com or bb284 [@] snu [dot] edu [dot] in.